Definition at line 32 of file mcljsimulation.h.
Public Member Functions | |
| MCLJ_Simulation (const ConfigurationDatabase &conf) | |
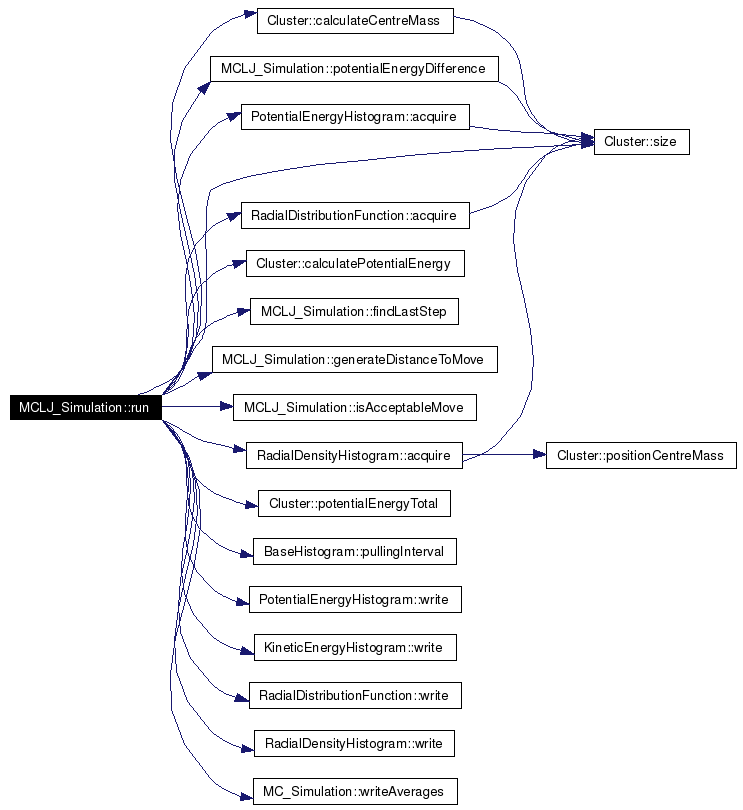
| virtual void | run (Cluster &cluster, string currentRun, string previousRun) |
Protected Member Functions | |
| virtual double | findLastStep (string previousRun) const |
Private Member Functions | |
| Coordinate | generateDistanceToMove () const |
| bool | isAcceptableMove (double potentialEnergyDifference) const |
| double | potentialEnergyDifference (const Cluster &cluster, const Atom &examinedAtom, const Coordinate &testPosition) const |
Private Attributes | |
| double | m_potentialEnergy |
|
|
General constructor for a Monte Carlo Lennard-Jones simulation.
Definition at line 36 of file mcljsimulation.cpp. References m_potentialEnergy. |
|
|
Finds the last step in the a series of simulations.
Implements BaseSimulation. Definition at line 142 of file mcljsimulation.cpp. Referenced by run(). |
|
|
This randomly generates a distance to move an atom.
Definition at line 155 of file mcljsimulation.cpp. References BaseSimulation::m_changeInStep, Coordinate::x, Coordinate::y, and Coordinate::z. Referenced by run(). |
|
|
This determines if a suggested move is acceptable based upon the potential energy difference
Definition at line 165 of file mcljsimulation.cpp. Referenced by run(). |
|
||||||||||||||||
|
Calculates the potential energy difference of moving one atom in a Cluster.
Definition at line 173 of file mcljsimulation.cpp. References Atom::properties, Cluster::size(), Coordinate::x, Coordinate::y, and Coordinate::z. Referenced by run(). Here is the call graph for this function:  |
|
||||||||||||||||
|
|
Definition at line 61 of file mcljsimulation.h. Referenced by MCLJ_Simulation(), and run(). |
 1.4.4
1.4.4